
SOTATERCEPT MSD FRANCE 45 mg, poudre et solvant pour solution injectable, boîte de 1 flacon de poudre seringue de solvant de 0,90 mL
Dernière révision : 04/06/2024
Taux de TVA : 0%
Laboratoire exploitant : MSD FRANCE
Source :

Sotatercept MSD France est indiqué pour le traitement de l'hypertension artérielle pulmonaire⃰ (HTAP) chez les adultes en classe fonctionnelle (CF) II ou III de l'OMS, recevant un traitement standard de l'HTAP en trithérapie incluant un antagoniste des récepteurs de l'endothéline (ARE), un inhibiteur de la phosphodiestérase 5 (iPDE5) ou un stimulateur de la guanylate cyclase soluble (GCs) et un analogue de la prostacycline par voie parentérale.
* L'efficacité a été montrée chez des patients présentant une HTAP incluant l'HTAP idiopathique, héritable, associée à une connectivite, induite par des médicaments ou des toxiques, ou associée à une cardiopathie congénitale corrigée (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Augmentation du taux d'hémoglobine
Des augmentations du taux d'Hb ont été observées chez des patients en cours de traitement par le sotatercept. Une polyglobulie sévère peut majorer le risque d'événements thromboemboliques et de syndrome d'hyperviscosité. Le taux d'Hb et le nombre d'hématies doit être contrôlé avant chaque administration jusqu'à ce qu'ils soient stables et périodiquement par la suite, afin de déterminer si des ajustements posologiques sont nécessaires (voir rubriques Posologie et mode d'administration et Effets indésirables).
Thrombocytopénie sévère
Une diminution du nombre de plaquettes a été observée chez certains patients traités par le sotatercept incluant une thrombocytopénie sévère (nombre de plaquettes < 50 x 109/L (<50 000/mm3)). Une thrombocytopénie est survenue plus fréquemment chez les patients recevant également une perfusion de prostacycline. Une thrombocytopénie sévère peut augmenter le risque de saignements (voirrubrique Effets indésirables). Le nombre de plaquettes doit être contrôlé avant la première administration, avant l'augmentation jusqu'à la dose cible, et périodiquement par la suite afin de déterminer si des ajustements posologiques sont nécessaires (voir rubriques Posologie et mode d'administration et Effets indésirables).
Saignement grave
Dans les études cliniques, des saignements graves (incluant des hémorragies gastro-intestinales et intracrâniennes) ont été observés chez 4,3 % des patients au cours du traitement par le sotatercept.
Les patients présentant des saignements graves étaient plus susceptibles d'être sous traitement de fond avec une prostacycline et/ou un antithrombotique ou d'avoir un faible nombre de plaquettes. Tout signe de saignements survenant en cours de traitement doit faire l'objet d'une surveillance avec contrôle des plaquettes sanguines. Le sotatercept ne doit pas être administré si le patient présente un saignement grave ou un nombre de plaquettes bas (voir rubrique Posologie et mode d'administration.).
Femme en âge de procréer
Le sotatercept peut nuire au foetus lorsqu'il est administré à une femme enceinte (voir rubriques Fertilité, grossesse et allaitement). Par conséquent, les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement par le sotatercept et jusqu'à au moins 4 mois après la dernière dose. Un test de grossesse est recommandé avant le début du traitement.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés étaient : céphalées (24,5 %), épistaxis (22,1 %), télangiectasies (16,6 %), diarrhée (15,3 %) et sensations vertigineuses (14,7 %).
Les effets indésirables graves les plus fréquemment rapportés étaient la thrombocytopénie (<1 %) et l'épistaxis (<1 %).
Les effets indésirables les plus fréquents ayant conduit à l'arrêt du traitement étaient l'épistaxis et la télangiectasie.
L'incidence globale des arrêts de traitement dus à un effet indésirable était de 4 % dans le groupe sotatercept et de 7 % dans le groupe placebo. Aucun effet indésirable spécifique entraînant l'arrêt du traitement et avec une fréquence supérieure à 1 % n'est survenu plus fréquemment dans le groupe de patients traités par le sotatercept.
Liste tabulée des effets indésirables
La sécurité du sotatercept a été évaluée dans l'étude pivot STELLAR, une étude contrôlée contre placebo menée chez 163 patients atteints d'HTAP traités par le sotatercept (voir rubrique Propriétés pharmacodynamiques). La durée médiane de traitement par le sotatercept était de 313 jours (environ 45 semaines).
Les effets indésirables rapportés avec Sotatercept MSD France sont répertoriés dans le tableau ci- dessous par classe de systèmes d'organes MedDRA et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000).
Tableau 4 : Effets indésirables
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Affections hématologiques et du système lymphatique | Très fréquent | Thrombocytopénie1,2 |
| Fréquent | Augmentation du tax d'hémoglobine1,3 | |
| Affections du système nerveux | Très fréquent | Sensations vertigineuses Céphalée |
| Affections respiratoires, thoraciques et médiastinales | Très fréquent | Epistaxis1 |
| Affections gastrointestinales | Très fréquent | Diarrhée |
| Affections de la peau et du tissu sous-cutané | Très fréquent | Télangiectasie1 Réactions au site d'injection (érythème, prurit, douleur) |
| Fréquent | Rash | |
| Investigations | Fréquent | Augmentation de artérielle1, 4 |
1 Voir la description des effets indésirables sélectionnés
2 Inclut « thrombocytopénie » et « diminution du nombre de plaquettes »
3 Inclut « augmentation du taux d'hémoglobine » et « polyglobulie »
4 Inclut « hypertension », « augmentation de la pression artérielle diastolique » et « augmentation de la pression artérielle »
Description des effets indésirables sélectionnés
Augmentation du taux d'hémoglobine
Une augmentation du taux d'Hb a été rapportée chez 8,6 % des patients traités par le sotatercept. D'après les données d'analyses biologiques, des augmentations modérées du taux d'Hb (>1,24 mmol/L (> 2 g/dL) au-dessus de la LSN) sont survenues chez 15,3 % des patients traités par le sotatercept. Les augmentations du taux d'Hb étaient gérées par des ajustements posologiques (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Thrombocytopénie
Une thrombocytopénie a été rapportée chez 10,4 % des patients traité par le sotatercept. Une réduction sévère du nombre de plaquettes < 50 x 109/L (< 50 000/mm3) est survenue chez 2,5 % des patients traités par le sotatercept. La thrombocytopénie était gérée par des ajustements posologiques (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Saignements graves :
Dans les études cliniques, des saignements graves (incluant des hémorragies gastro-intestinales et intracrâniennes) ont été observés chez 4,3 % des patients au cours du traitement par sotatercept.
Les patients présentant des saignements graves étaient plus susceptibles d'être sous traitement de fond avec une prostacycline et/ou un antithrombotique ou d'avoir un faible nombre de plaquettes. Tout signe de saignements survenant en cours de traitement doit faire l'objet d'une surveillance avec contrôle des plaquettes sanguines. Le sotatercept ne doit pas être administré si le patient présente un saignement grave ou un nombre de plaquettes bas (voir rubrique Posologie et mode d'administration.).
Télangiectasie
Des télangiectasies ont été observées chez 16,6 % des patients traités par le sotatercept. Le délai médian d'apparition était de 47,1 semaines. Les arrêts de traitement dus à des télangiectasies étaient de 1 % dans le groupe sotatercept.
Augmentation de la pression artérielle
Une augmentation de la pression artérielle a été rapportée chez 4,3 % des patients prenant Sotatercept. Chez les patients prenant Sotatercept, la pression artérielle systolique moyenne a augmenté de 2,2 mm Hg par rapport à l'inclusion et la pression artérielle diastolique moyenne a augmenté de 4,9 mm Hg en 24 semaines. Chez les patients prenant le placebo, la pression artérielle systolique moyenne a diminué de 1,6 mm Hg par rapport à l'inclusion et la pression artérielle diastolique moyenne a diminué de 0,6 mm Hg en 24 semaines.
Personnes âgées
A l'exception des événements hémorragiques, aucune différence n'a été observée en terme de sécurité entre le sous-groupe des personnes âgées de < 65 ans et le sous-groupe des personnes âgées de ≥ 65 ans. Les événements hémorragiques sont survenus plus fréquemment dans le sous-groupe des patients de plus de 65 ans traités par le sotatercept (52 %) comparativement aux patients de < 65 ans (31,9 %). Il n'a pas été observé de déséquilibre notable pour un événement hémorragique spécifique entre les différentes catégories d'âge
Données de sécurité à long terme
Les données de sécurité à long terme décrites proviennent des données issues du regroupement des études cliniques de phase II, de phase III et de suivi en ouvert (n=431). Dans cette analyse, la durée médiane d'exposition était de 657 jours (environ 94 semaines) avec 343 patients ayant atteint au moins 52 semaines d'exposition. Le profil de sécurité était généralement similaire à celui observé dans l'étude pivot STELLAR avec une augmentation de l'incidence rapportée des épisodes de saignements (dont 7% d'hémorragies graves) par rapport à la période d'observation dans l'étude STELLAR, ce qui témoigne de la nécessité, en pratique, d'une surveillance du taux de plaquettes tout au long du traitement .Voir rubrique Posologie et mode d'administration.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
- SURVEILLANCE du traitement :
- - Taux d'Hb et nombre d'hématies : à contrôler avant chaque administration jusqu'à ce qu'ils soient stables puis de façon périodique.
- - Nombre de plaquettes : à contrôler avant la première administration, avant l'augmentation jusqu'à la dose cible, et périodiquement par la suite.
- FEMME en âge de PROCREER :
- - Il est recommandé de faire un test de grossesse avant le début du traitement.
- - Utiliser une méthode de contraception efficace pendant le traitement et jusqu'à au moins 4 mois après la dernière dose.
- ALLAITEMENT : NE PAS allaiter pendant le traitement et pendant 4 mois après la dernière dose du traitement.
Femmes en âge de procréer
Un test de grossesse est recommandé chez les femmes en âge de procréer avant le début du traitement. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par le sotatercept et jusqu'à au moins 4 mois après la dernière dose en cas d'arrêt du traitement (voir rubriques Mises en garde spéciales et précautions d'emploi).
Grossesse
Il n'existe pas de données sur l'utilisation du sotatercept chez la femme enceinte. Les études menées chez l'animal ont mis en évidence une toxicité sur la reproduction (pertes post-implantatoires ou toxicité foetale) (voir rubriques Données de sécurité préclinique).
Sotatercept n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
On ne sait pas si le sotatercept/ses métabolites sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
L'allaitement doit être arrêté pendant le traitement par le sotatercept et repris 4 mois après la dernière dose du traitement.
Fertilité
Compte tenu des observations chez l'animal, le sotatercept peut altérer la fertilité féminine et masculine (voir rubriques Mises en garde spéciales et précautions d'emploi et Données de sécurité préclinique). Les effets indésirables sur la fertilité des rats mâles étaient réversibles après une période de 13 semaines.
Aucune étude d'interaction n'a été réalisée.
Le traitement par sotatercept doit être initié et supervisé uniquement par un médecin expérimenté dans le diagnostic et le traitement de l'HTAP.
Posologie
Sotatercept MSD France est administré une fois toutes les 3 semaines en une seule injection sous- cutanée. La dose à administrer est déterminée en fonction du poids corporel du patient (voir Tableaux 1 et 2 ci-dessous).
Dose initiale recommandée
Le taux sanguin d'hémoglobine (Hb) et le nombre de plaquettes doivent être évalués avant l'administration (le jour même ou au maximum 3 jours avant). Il n'est pas recommandé d'initier le traitement si le nombre de plaquettes est < 50 x 109/L (< 50 000/mm3), voir rubrique Mises en garde spéciales et précautions d'emploi.
Le traitement doit être débuté à la dose initiale de 0,3 mg/kg (voir Tableau 1 ci-dessous).
Tableau 1 : Volume de la solution reconstituée à injecter pour une dose de 0,3 mg/kg
| Intervalle de poids corporel du patient (kg) | Volume de solution reconstituée à injecter (mL)* | Type de kit |
| 30,0-40,8 | 0,2 | Kit contenant 1 flacon de 45 mg |
| 40,9-57,4 | 0,3 | |
| 57,5-74,1 | 0,4 | |
| 74,2-90,8 | 0,5 | |
| 90,9-107,4 | 0,6 | |
| 107,5-124,1 | 0,7 | |
| 124,2-140,8 | 0,8 | |
| 140,9-157,4 | 0,9 | |
| 157,5-174,1 | 1,0 | Kit contenant 1 flacon de 60 mg |
| 174,2-180,0 | 1,1 |
*La concentration de la solution reconstituée est de 50 mg/mL de sotatercept MSD France (voir rubrique Précautions particulières d'élimination et de manipulation)
Dose cible recommandée
Après avoir vérifié que le taux d'Hb est acceptable (c'est-à-dire un taux resté à moins de 1,24 mmol/L, (2 g/dL) au-dessus de la valeur de base du patient) et que le nombre de plaquettes est supérieur à 50 x 109/L (> 50 000/mm3) (voir rubrique Mises en garde spéciales et précautions d'emploi), la dose sera augmentée à 0,7 mg/kg. Le traitement doit être poursuivi à la dose de 0,7 mg/kg toutes les 3 semaines, sauf si des ajustements posologiques sont nécessaires (voir Tableau 3).
Tableau 2 : Volume de la solution reconstituée à injecter pour une dose de 0,7 mg/kg
| Intervalle de poids corporel du patient (kg) | Volume de solution reconstituée à injecter (mL)* | Type de kit |
| 30,0-31,7 | 0,4 | Kit contenant 1 flacon de 45 mg |
| 31,8-38,9 | 0,5 | |
| 39,0-46,0 | 0,6 | |
| 46,1-53,2 | 0,7 | |
| 53,3-60,3 | 0,8 | |
| 60,4-67,4 | 0,9 | |
| 67,5-74,6 | 1,0 | Kit contenant 1 flacon de 60 mg |
| 74,7-81,7 | 1,1 | |
| 81,8-88,9 | 1,2 | |
| 89,0-96,0 | 1,3 | 2 kits contenant chacun 1 flacon de 45 mg |
| 96,1-103,2 | 1,4 | |
| 103,3-110,3 | 1,5 | |
| 110,4-117,4 | 1,6 | |
| 117,5-124,6 | 1,7 | |
| 124,7-131,7 | 1,8 | |
| 131,8-138,9 | 1,9 | 2 kits contenant chacun 1 flacon de 60 mg |
| 139,0-146,0 | 2,0 | |
| 146,1-153,2 | 2,1 | |
| 153,3-160,3 | 2,2 | |
| 160,4-167,4 | 2,3 | |
| 167,5 et plus | 2,4 |
*La concentration de la solution reconstituée est de 50 mg/mL de sotatercept MSD France (voir rubrique Précautions particulières d'élimination et de manipulation)
Ajustements posologiques en raison d'une augmentation du taux d'hémoglobine ou d'une diminution dunombre de plaquettes
Des augmentations du taux d'Hb à des valeurs supérieures à 1,24 mmol/L (2 g/dL) au-dessus de la limite supérieure de la normale (LSN) et une diminution du nombre de plaquettes à < 50 x 109/L (< 50 000/mm3) ont été observées avec le sotatercept. Le taux d'Hb et le nombre de plaquettes doivent être contrôlés tout au long du traitement et la dose doit être ajustée si nécessaire (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables). Consultez le Tableau 3 pour les instructions de modification posologique.
Tableau 3 : Modifications posologiques
| SotaterceptRecommandations posologiques | |
| Dose d'initiation | |
| Traitement initial | 0,3 mg/kg |
| Augmentation à la dose cible | |
| Si le taux d'Hb reste à moins de 1,24 mmol/L (2 g/dL) au-dessus de la valeur de base du patient et le nombre de plaquettes est > 50.0 x 109/L (> 50 000/mm3) | Augmenter la dose à 0,7 mg/kg toutes les 3 semaines |
| Modifications posologiques en raison de l'augmentation du taux d'Hb | |
| Si le taux d'Hb augmente de plus de 1,24 mmol/L (2,0 g/dL) au-dessus de la valeur précédente et le taux d'Hb est au-dessus de la LSN | Reporter le traitement de 3 semaines. |
| Après un report de traitement, si le taux d'Hb est inférieur à 1,24 mmol/L (2,0 g/dL) au-dessus de la valeur initiale ou le taux d'Hb est inférieur à la LSN | Reprendre le traitement à la dose précédentePour les patients qui étaient précédemment à la dose de 0,3 mg/kg : après 2 doses consécutives à 0,3 mg/kg, la dose peut être augmentée à la dose cible de 0,7 mg/kg |
| Si le taux d'Hb augmente de > 2,48 mmol/L (4,0 g/dL) au-dessus de la valeur de base du patient et le taux d'Hb est au-dessus de la LSN | L'arrêt du traitement doit être envisagé |
| Prolongation du report de traitement en raison de l'augmentation du taux d'Hb | |
| Si le traitement est reporté plus de 9 semaines (3 reports consécutifs) | Reprendre le traitement à la dose de 0,3 mg/kg.Si le patient est déjà à la dose réduite de 0,3 mg/kg, l'arrêt du traitement doit être envisagé |
| Modifications posologiques en raison de diminutions du nombre de plaquettes | |
| Nombre de plaquettes < 50 000/mm3 (< 50.0 x 109/L) | Reporter le traitement de 3 semaines. |
| Après un report de traitement, si le nombre de plaquettes est > 50 000/mm3 (> 50.0 x 109/L) | Envisager de reprendre le traitement à la dose de 0,3 mg/kgPour les patients qui étaient précédemment à la dose de 0,3 mg/kg : après 2 doses consécutives à 0,3 mg/kg, la dose peut être augmentée à la dose cible de 0,7 mg/kg |
| Prolongation du report de traitement en raison de diminutions du nombre de plaquettes | |
| Si le traitement est reporté plus de 9 semaines (3 reports consécutifs) | L'arrêt du traitement doit être envisagé |
Conduite à tenir en cas de télangiectasie
En cas d'identification de nouveaux événements de télangiectasie, de gravité/intensité modérée ou élevée, ou en cas de progression légère à modérée de télangiectasie, l'injection doit être reportée de 3
semaines (un report de dose) si le patient recevait une dose de 0,7 mg/kg, ou de 9 semaines (3 reports consécutifs) si le patient recevait une dose de 0,3 mg/kg au moment de l'événement. Si après le(s) report(s) de dose(s), il n'y a eu aucune aggravation de l'événement de télangiectasie, le traitement peut être repris à une dose de 0,3 mg/kg. Si l'événement de télangiectasie progresse pendant la période du report de dose, l'arrêt du traitement doit être envisagé.
Dose oubliée
En cas d'oubli d'une dose de sotatercept, celle-ci doit être administrée dès que possible. Si la dose oubliée de sotatercept n'est pas administrée dans les 3 jours suivant la date prévue, ajuster le schéma posologique afin de maintenir des intervalles de 3 semaines entre les administrations.
Populations particulières
Personnes âgées
Aucun ajustement posologique n'est nécessaire chez les patients âgés de ≥ 65 ans (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire en cas d'insuffisance rénale. Le sotatercept n'a pas été étudié chez les patients atteints d'HTAP présentant une insuffisance rénale sévère (DFGe < 30 mL/min/1,73 m2) (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire en cas d'insuffisance hépatique. L'insuffisance hépatique ne devrait pas influencer le métabolisme du sotatercept dans la mesure où il est métabolisé par catabolisme intracellulaire. Le sotatercept n'a pas été étudié chez les patients présentant une insuffisance hépatique (classification de Child-Pugh A à C) (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du sotatercept chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
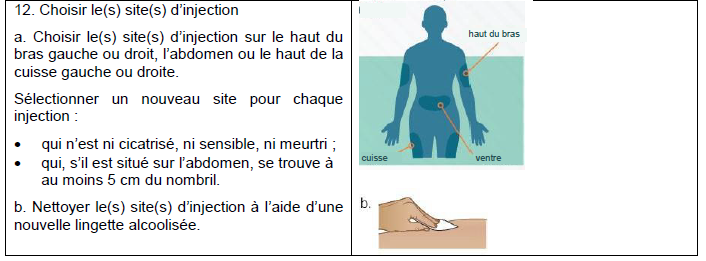
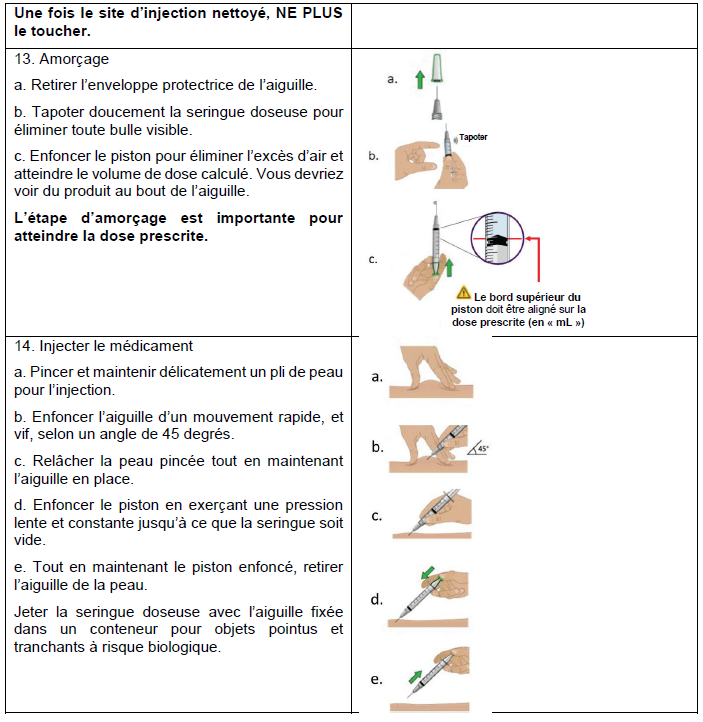
Sotatercept MSD France doit être reconstitué avant utilisation et administré par injection sous-cutanée au niveau de l'abdomen (à au moins 5 cm du nombril), le haut du bras ou de la cuisse.
Voir la rubrique Précautions particulières d'élimination et de manipulation et les Instructions pour l'utilisation pour des instructions détaillées sur la préparation et l'administration appropriées du sotatercept.
L'injection de Sotatercept MSD France doit être réalisée par un professionnel de santé.
Durée de conservation :
3 ans
Apres reconstitution :
- la stabilité physicochimique de la solution reconstituée dans le flacon d'origine a été démontrée pendant 24 heures entre 2°C et 8°C incluant un maximum de 8 heures à température ambiante.
- la stabilité physicochimique de la solution reconstituée dans la seringue pour administration a été démontrée pendant 4 heures à température ambiante
D'un point de vue microbiologique, sauf si la méthode de reconstitution prévient tout risque de contamination microbienne, le médicament doit être utilisé immédiatement.
En cas d'utilisation non immédiate, les durées et conditions de conservation après reconstitution et avant utilisation relèvent de la responsabilité de l'utilisateur.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
A conserver dans l'emballage extérieur d'origine, à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments, à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
Chez les volontaires sains, le sotatercept administré à la dose de 1 mg/kg a entraîné une augmentation du taux d'Hb associée à une hypertension. En cas de surdosage, surveiller étroitement les augmentations du taux d'Hb et de la pression artérielle ainsi que le nombre de plaquettes, et dispenser les soins appropriés. Le sotatercept n'est pas dialysable par hémodialyse.
Classe pharmacothérapeutique : {classe}, code ATC : {code} non encore attribué.
Mécanisme d'action
Le sotatercept est un inhibiteur de la signalisation de l'activine avec une sélectivité élevée pour l'activine A, une glycoprotéine dimérique qui appartient à la superfamille de ligands du facteur de croissance transformant β (TGF-β). L'activine A se lie au récepteur de l'activine de type IIA (ActRIIA) qui régule la signalisation clé de l'inflammation, de la prolifération cellulaire, de l'apoptose et de l'homéostasie tissulaire.
Les taux d'activine A sont augmentés chez les patients atteints d'HTAP. La liaison de l'activine à l'ActRIIA favorise la signalisation proliférative tandis que la signalisation antiproliférative du récepteur de la protéine morphogénétique osseuse de type II (BMPRII) diminue. Le déséquilibre de la signalisation ActRIIA-BMPRII sous-jacent à l'HTAP entraîne une hyperprolifération des cellules vasculaires, induisant un remodelage pathologique de la paroi artérielle pulmonaire à l'origine de l'HTAP.
Le sotatercept est une protéine de fusion homodimérique recombinante du récepteur de l'activine de type IIA-Fc (ActRIIA-Fc), qui agit comme un piège à ligands séquestrant l'excès d'activine A et d'autres ligands d'ActRIIA afin d'inhiber la signalisation de l'activine. En conséquence, le sotatercept rééquilibre la signalisation pro-proliférative (médiée par ActRIIA/Smad2/3) et antiproliférative (médiée par BMPRII/Smad1/5/8), pour moduler la prolifération vasculaire.
Effets pharmacodynamiques
Dans des modèles d'HTAP chez le rat, un analogue du sotatercept a réduit l'expression des marqueurs pro-inflammatoires au niveau de la paroi artérielle pulmonaire, a réduit le recrutement de leucocytes, a inhibé la prolifération des cellules endothéliales et des cellules musculaires lisses et a favorisé leur apoptose dans le système vasculaire malade. Ces modifications cellulaires étaient associées à un amincissement de la paroi des vaisseaux, à une inversion du remodelage artériel et ventriculaire droit et à une amélioration de l'hémodynamique.
Une étude clinique de phase II a évalué les résistances vasculaires pulmonaires (RVP) chez des patients atteints d'HTAP après 24 semaines de traitement par le sotatercept. La diminution des RVP par rapport à l'inclusion était significativement plus importante dans les groupes sotatercept 0,7 mg/kg et 0,3 mg/kg que dans le groupe placebo. La différence moyenne des moindres carrés (MC) ajustée au placebo par rapport à l'inclusion était de -269,4 dynes*s/cm5 (IC à 95 % : -365,8 ; -173,0) pour le groupe sotatercept 0,7 mg/kg et de -151,1 dynes*s/cm5 (IC à 95 % : -249,6 ; -52,6) pour le groupe sotatercept 0,3 mg/kg. Dans l'étude STELLAR, la diminution des RVP par rapport à l'inclusion était également significativement plus importante dans le groupe sotatercept 0,7 mg/kg par rapport au groupe placebo (voir « Efficacité et sécurité clinique ci-dessous»).
Efficacité et sécurité clinique
L'efficacité du sotatercept a été évaluée chez des patients adultes atteints d'HTAP dans l'étude pivot STELLAR. STELLAR était une étude clinique, en double aveugle, contrôlée contre placebo, multicentrique et en groupes parallèles, dans laquelle 323 adultes atteints d'HTAP (groupe 1 de l'OMS) en classe fonctionnelle II ou III ont été randomisés selon un rapport de 1/1 pour recevoir du sotatercept à la dose initiale de 0,3 mg/kg augmentée jusqu'à la dose cible de 0,7 mg/kg (n = 163) ou le placebo (n = 160) administré par voie sous-cutanée une fois toutes les 3 semaines. Les patients ont continué leur traitement au cours de la période de traitement à long terme en double aveugle jusqu'à ce que tous les patients aient terminé la semaine 24.
L'âge médian des patients inclus était de 48,0 ans (18 à 82 ans), parmi lesquels 16,7 % étaient âgés de ≥ 65 ans et 79,3 % étaient des femmes. Les étiologies de l'HTAP étaient idiopathique (58,5 %, n = 189), héritable (18,3 %, n = 59), associée à une connectivite (14,9 %, n = 48), associée à une cardiopathie congénitale avec shunts corrigée (5 %, n = 16) et celle induite par l'utilisation de toxiques ou de certains médicaments (3,4 %, n = 11). La durée moyenne entre le diagnostic d'HTAP et la sélection était de 8,76 ans. L'essai STELLAR a exclu les patients avec un diagnostic d'HTAP associée au virus de l'immunodéficience humaine (VIH), d'HTAP associée à une hypertension portale, d'HTAP associée à une schistosomiase et de maladie veino-occlusive pulmonaire. IL n'y a pas de données disponibles dans les HTAP ayant ces étiologies..
Les participants recevaient un traitement standard de l'HTAP, pour la plupart en trithérapie (61,3 %, n = 198) ou en bithérapie (34,7 %, n = 112). Plus d'un tiers (39,9 %, n= 129) recevait des perfusions de prostacycline, et 33,7% (n = 109) recevaient une trithérapie incluant un analogue parentéral de prostacycline. Les proportions de participants en classe en fonctionnelle II de l'OMS étaient de 48,6 % et en classe fonctionnelle III de l'OMS étaient de 51,4 %.
Le critère principal d'évaluation de l'efficacité était la distance parcourue au test de marche de 6 minutes (TM6) de l'inclusion à la Semaine 24. Dans le groupe traité par le sotatercept de la population globale de l'étude, la variation médiane de la distance parcourue au TM6 ajustée au placebo de l'inclusionà la Semaine 24 était de +40,8 mètres (IC à 95 % : 27,5, 54,1 ; p < 0,001). La variation médiane de la distance parcourue au TM6 corrigée par rapport au placebo de l'inclusion à la Semaine 24 a également été évaluée dans des sous-groupes. L'effet du traitement était cohérent parmi les différents sous- groupes prédéfinis, incluant le sexe, le 'étiologie de l'HTAP, le traitement de fond de l'HTAP à l'inclusion, le traitement par perfusion de prostacycline à l'inclusion, la classe fonctionnelle de l'OMS et les RVP à l'inclusion.
La progression de la maladie a été mesurée par le délai jusqu'à la survenue d'un premier événement d'aggravation clinique ou un décès. Les événements d'aggravation clinique étaient définis en : une inscription sur liste d'attente de transplantation pulmonaire et/ou cardiaque liée à une aggravation, la nécessité d'initier un traitement de secours avec un traitement approuvé de l'HTAP ou la nécessité d'augmenter la dose de prostacycline en perfusion de ≥ 10 %, la nécessité d'une atrioseptostomie, une hospitalisation pour aggravation de l'HTAP (≥ 24 heures) ou une détérioration de l'HTAP (aggravation de la classe fonctionnelle de l'OMS et diminution de la distance parcourue au TM6 de ≥ 15 %, les deux événements survenant en même temps ou à distance l'un de l'autre). Les événements d'aggravation clinique et les décès ont été enregistrés jusqu'à ce que le dernier patient ait effectué la visite de la Semaine 24 (données disponibles jusqu'à la date d'arrêt de collecte des données correspondant à une durée d'exposition médiane de 33,6 semaines).
Les patients traités par le sotatercept ont présenté une amélioration statistiquement significative de la classe fonctionnelle de l'OMS, et une réduction du risque d'événement d'aggravation clinique, y compris de décès et d'hospitalisation par rapport aux patients sous placebo.
La différence médiane de traitement de RVP entre le groupe traité par le sotatercept et le groupe placebo était de -234,6 dynes*sec/cm5 (IC à 95% : -288,4, -180,8 ; p<0,001). La différence médiane de traitement du NT-proBNP entre les groupes sotatercept et placebo était de -441,6 pg/mL (IC à 95 %: - 573,54, -309,61 ; p<0,001). Une amélioration de la classe fonctionnelle par rapport à l'inclusion a été observée chez 29 % des patients sous sotatercept contre 13,8 % sous placebo (p<0,001).
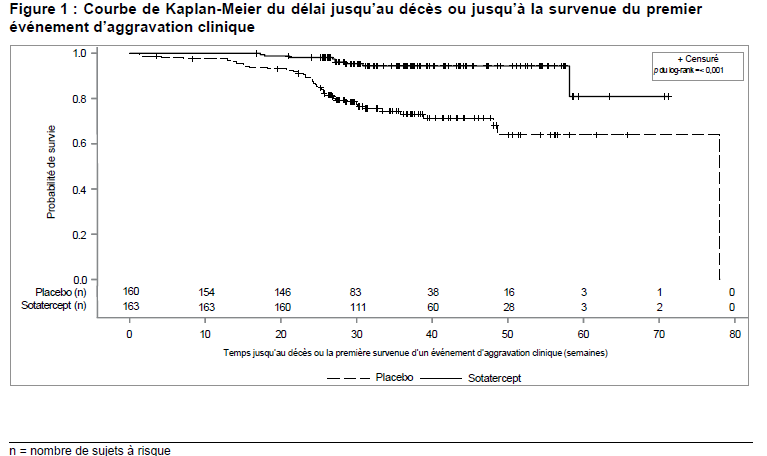
Le traitement par le sotatercept a entraîné une réduction de 82 % (HR 0,182, IC à 95% 0,075, 0,441 ; p<0,001) du risque de survenue d'un événement d'aggravation clinique ou d'un décès par rapport au placebo (voir Tableau 5 et Figure 1).
Tableau 5 : Décès ou événement d'aggravation clinique
| | Placebo (N = 160) | Sotatercept (N = 163) |
| Nombre total de patients décédés ou ayant présenté au moins un événement d'aggravation clinique, n (%) | 29 (18,1) | 7 (4,3) |
| Evaluation de la survenue du premier événement d'aggravation clinique* ou du décès n (%) | | |
| Décès | 6 (3,8) | 2 (1,2) |
| Inscription sur liste de transplantation pulmonaire et/ou cardiaque liée à une aggravation | 1 (0,6) | 1 (0,6) |
| Nécessité d'une septostomie auriculaire | 0 (0,0) | 0 (0,0) |
| Hospitalisation liée à l'HTAP (≥ 24 heures) | 8 (5,0) | 0 (0,0) |
| Détérioration de l'HTAP† | 15 (9,4) | 4 (2,5) |
* Un patient peut avoir plus d'une évaluation enregistrée pour leur premier événement d'aggravation clinique. Il y a eu deux patients recevant le placebo et aucun patient recevant le sotatercept qui ont eu plus d'une évaluation enregistrée pour leur premier événement d'aggravation clinique. Cette analyse excluait la composante « nécessité d'initier un traitement de secours avec un traitement approuvé de l'HTAP ou nécessité d'augmenter la dose de prostacycline en perfusion de 10 % ou plus ».
† La détérioration de l'HTAP est définie par la survenue des deux événements suivants à tout moment, même s'ils ont commencé à des moments différents, par rapport à leurs valeurs à l'inclusion : (a) aggravation de la classe fonctionnelle de l'OMS (II à III, III à IV, II à IV, etc.) ; (b) diminution de la distance parcourue au TM6 de ≥ 15 % (confirmée par deux TM6 à au moins 4 heures d'intervalle mais sans dépasser une semaine).
N = nombre de patients dans la population FAS ; n = nombre de patients dans la catégorie. Les pourcentages sont calculés sous forme de (n/N)*100.
![]()
La majorité des participants traités jusqu'à la semaine 24 dans l'étude STELLAR ont bien toléré le schéma posologique et n'ont nécessité ni retard de dose (92,6 %) ni réduction de dose (93,9 %). L'incidence globale des arrêts de traitement dus à un effet indésirable était de 4 % dans le groupe sotatercept et de 7 % dans le groupe placebo.
Effets dans la population des patients recevant une trithérapie incluant un analogue parentéral de laprostacycline à l'inclusion
Un bénéfice clinique a été observé chez les patients recevant une trithérapie incluant un analogue parentéral de la prostacycline à l'inclusion, qui représentaient 33,7% (n = 109) de la population totale de l'étude, avec :
- une amélioration de la capacité fonctionnelle comprenant à la fois une amélioration de la distance parcourue au TM6 (+42,8 mètres, IC95% [21,18 ; 64,45], p<0,001), de la classe fonctionnelle de l'OMS (placebo 10,7% contre sotatercept 32,1%, p<0,012 ; avec environ trois fois plus de patients améliorant leur classe fonctionnelle de III à II (9,8% vs 26,9%) et de II à I (2,0% vs 5,8%)) à la semaine 24,
- une réduction de 85% du risque de morbi-mortalité, après un suivi médian de 37,3 semaines (HR : 0,149, IC95% [0,043 ; 0,514], p<0,001), et après un suivi médian de 45,9 semaines (HR : 0,149, IC95% [0,043 ; 0,516], p<0,001).
- une amélioration des paramètres hémodynamiques et des biomarqueurs comprenant une diminution des RVP (-271,6 dynes*sec/cm5, IC95% [-366,35 ; -176,84], p<0,001) et du taux de NT-proBNP (-396,0 pg/mL, IC95% [-635,65 ; -156,36], p<0,001) à la semaine 24.
Immunogénicité
L'incidence observée d'anticorps anti-médicament dépend fortement de la sensibilité et de la spécificité de la méthode de dosage. Les différences dans les méthodes de dosage utilisées empêchent toute comparaison significative de l'incidence des anticorps anti-médicament dans l'étude décrite ci-dessous avec l'incidence des anticorps anti-médicament dans d'autres études, y compris des études portant sur le sotatercept ou d'autres produits à base de sotatercept.
Pendant la période de traitement de 24 semaines dans l'étude pivot (STELLAR), 42/162 (25,9 %) des patients traités par le sotatercept ont développé des anticorps anti-sotatercept. Parmi ces 42 patients, 11 (6,8 % de tous les patients recevant du sotatercept) ont été testés positifs aux anticorps neutralisants dirigés contre le sotatercept. Les taux d'anticorps anti-sotatercept étaient généralement faibles avec un taux médian de 30 (intervalle : < 20 à 640).
Aucun effet clinique des anticorps anti-sotatercept n'a été identifié sur la pharmacocinétique, la pharmacodynamie, la sécurité ou l'efficacité du sotatercept pendant la durée du traitement de 24 semaines.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec le sotatercept dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de l'hypertension artérielle pulmonaire (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Chez les patients atteints d'HTAP, la moyenne géométrique (coefficient de variation en % [CV %]) de l'ASC à l'état d'équilibre et de la concentration maximale (Cmax) à l'état d'équilibre, à la dose de 0,7 mg/kg toutes les 3 semaines, était respectivement de 171,3 mcg×j/mL (34,2 %) et 9,7 mcg/mL (30 %). L'ASC et la Cmax du sotatercept augmentent proportionnellement à la dose. L'état d'équilibre est atteint après environ 15 semaines en cas d'administrations multiples toutes les 3 semaines. Le rapport d'accumulation de l'ASC du sotatercept était d'environ 2,2.
Absorption
La formulation sous-cutanée (SC) a une biodisponibilité absolue d'environ 66 %. La concentration maximale du sotatercept est atteinte en un temps médian jusqu'à la concentration maximale du médicament (Tmax) d'environ 7 jours (intervalle de 2 à 8 jours) après des doses sous cutanées multiples (0,1 mg/kg toutes les 4 semaines) chez les femmes ménopausées.
Distribution
Le volume de distribution centrale (CV %) du sotatercept est d'environ 3,6 L (24,7 %). Le volume de distribution périphérique (CV %) est d'environ 1,7 L (73,3 %).
Biotransformation
Le sotatercept est catabolisé par des processus généraux de dégradation des protéines.
Elimination
La clairance du sotatercept est d'environ 0,18 L/jour. La moyenne géométrique de la demi-vie terminale (CV %) est d'environ 21 jours (33,8 %).
Populations particulières
Age, sexe et origine ethnique
Aucune différence cliniquement significative de la pharmacocinétique (PK) du sotatercept n'a été observée en fonction de l'âge (18 à 81 ans), du sexe ou de l'origine ethnique.
Poids corporel
La clairance et le volume de distribution centrale du sotatercept ont augmenté avec le poids corporel. Le schéma posologique recommandé en fonction du poids entraine des expositions constantes du sotatercept, quel que soit le poids corporel.
Insuffisance rénale
La pharmacocinétique du sotatercept était comparable chez les patients atteints d'HTAP présentant une insuffisance rénale légère à modérée (DFGe compris entre 30 et 89 mL/min/1,73 m2) à ceux ayant une fonction rénale normale (DFGe ≥ 90 mL/min/1,73 m2). De plus, la pharmacocinétique du sotatercept est comparable entre les patients atteints d'insuffisance rénale terminale (IRT) sans HTAP et les patients présentant une fonction rénale normale. Sotatercept n'est pas dialysable pendant l'hémodialyse. Aucun ajustement posologique n'est recommandé chez les patients insuffisants rénaux. Le sotatercept n'a pas été étudié chez les patients atteints d'HTAP présentant une insuffisance rénale sévère (DFGe< 30 mL/min/1,73 m 2).
Insuffisance hépatique
L'insuffisance hépatique (déterminée par la classification de Child-Pugh) ne devrait pas influencer le métabolisme du sotatercept puisqu'il est métabolisé par catabolisme intra-cellulaire. Le sotatercept n'a pas été étudié chez les patients atteints d'HTAP présentant une insuffisance hépatique (Classification Child-Pugh A à C).
Il n'a pas été réalisé d'étude sur la conduite avec le sotatercept.
A priori, le sotatercept n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Aucune étude de carcinogénicité ou de mutagénicité n'a été menée avec le sotatercept.
Toxicité à doses répétées
Chez les rats et les singes, les études de toxicité par voie sous cutanée les plus longues ont duré respectivement 3 mois et 9 mois. Chez les rats recevant des doses hebdomadaires de 0,3, 3 et 30 mg/kg pendant 3 mois, les effets toxiques observés étaient une dégénérescence du canal déférent/testiculaire, une congestion/nécrose de la glande surrénale ainsi qu'une glomérulonéphrite membrano-proliférative et une néphrite tubulo-interstitielle au niveau des reins survenues à une exposition 18 fois supérieure à la dose humaine maximale recommandée (DHMR) (sur la base de l'aire sous courbe (ASC) estimée). Les modifications surrénales et rénales étaient réversibles après une période de récupération de 1 mois. Chez des singes recevant des doses de 1, 2,6 et 10 mg/kg une fois toutes les 4 semaines et de 10 mg/kg une fois toutes les 2 semaines, les effets toxiques étaient limités à une glomérulonéphrite et une néphrite tubulo-interstitielle au niveau des reins survenues à des expositions ≥ 6 fois la DHMR (sur la base de l'ASC estimée). Les modifications rénales chez les singes étaient partiellement résolues après une période de récupération de 3 mois.
Toxicité sur la reproduction
Dans une étude de fertilité et de développement embryonnaire précoce chez les rates, le sotatercept a été administré par voie sous cutanée une fois par semaine à des doses de 5, 15 et 50 mg/kg pendant 2 semaines avant l'accouplement et jusqu'au Jour 7 de la gestation. Aux doses ≥ 15 mg/kg (≥ 9 fois la DHMR, sur la base de l'ASC estimée), les taux de gestation étaient diminués et des augmentations des pertes pré-implantatoires et post-implantatoires ainsi que des réductions de la taille des portées vivantes étaient observées. L'augmentation de la durée du cycle oestral était observée à la dose de 50 mg/kg uniquement (21 fois la DHMR, sur la base de l'ASC estimée).Dans une étude de fertilité chez les rats, le sotatercept a été administré par voie sous cutanée une fois par semaine à des doses de 0,3, 3 et 30 mg/kg pendant 13 semaines (débutant 10 semaines avant l'accouplement). Un sous-groupe d'animaux a été examiné après une période de récupération de 13 semaines. Aux doses ≥ 0,3 mg/kg (0,5 fois la DHMR, sur la base de l'ASC estimée), des modifications histologiques irréversibles étaient observées au niveau des canaux déférents, des testicules et des épididymes. Des diminutions réversibles de la fertilité étaient observées à la dose de 30 mg/kg (20 fois la DHMR, sur la base de l'ASC estimée).
Dans les études de toxicité sur le développement embryo-foetal, le sotatercept a été administré par voie SC à des animaux gravides pendant la période d'organogenèse. Sotatercept a été administré à des rates aux Jours 6 et 13 de la gestation à des doses de 5, 15 ou 50 mg/kg et à des lapines aux Jours 7 et 14 de la gestation à des doses de 0,5, 1,5 ou 5 mg/kg. Les effets observés chez les deux espèces étaient notamment des réductions des nombres de foetus vivants et des poids corporels des foetus, des retards d'ossification et des augmentations des résorptions et des pertes post-implantatoires. Chez les rates et les lapines, ces effets étaient observés à des expositions (sur la base de l'ASC) d'environ respectivement 4 fois et 0,6 fois la DHMR. Chez les rates uniquement, des variations squelettiques (augmentation du nombre de côtes surnuméraires et modifications du nombre de vertèbres thoraciques ou lombaires) sont survenues à une exposition 15 fois supérieure à l'exposition humaine à la DHMR.
Dans une étude de développement pré et postnatal chez les rates, le sotatercept a été administré par voie SC à des doses de 1,5 et 5 mg/kg aux Jours 6 et 13 de la gestation ou à des doses de 1,5, 5 ou 10 mg/kg pendant l'allaitement aux Jours 1, 8 et 15. Aucun effet indésirable n'a été observé chez les petits de la première génération (F1) dont les mères étaient traitées par le sotatercept pendant la gestation à des expositions estimées jusqu'à 2 fois la DHMR. Chez les petits F1 dont les mères étaient traitées pendant l'allaitement, les diminutions du poids des petits étaient corrélées à des retards de maturation sexuelle à des expositions estimées (sur la base de l'ASC) à ≥ 2 fois la DHMR.


Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à prescription hospitalière réservée aux spécialistes en pneumologie, cardiologie ou en médecine interne.
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament nécessitant une surveillance particulière pendant le traitement.
Poudre et solvant pour solution injectable.
Poudre : poudre blanche à blanc cassé.
Solvant : solution limpide et incolore.
45 mg de poudre en flacon (verre de type I)
muni d'un bouchon en bromobutyle et un opercule (aluminium avec une
capsule amovible de type flip-off en polypropylène vert) ; 1 mL de
solvant en seringue préremplie (cartouche en verre de type I fermée par
un bouchon en caoutchouc bromobutyle)
Sotatercept MSD France se présente en :
· Kit contenant 1 flacon de poudre, 1 seringue préremplie de solvant, 1 seringue doseuse graduée au dixième de millilitre (0,1 mL), 1 dispositif de transfert (13 mm), 1 aiguille d'injection et 3 tampons alcoolisés.
Chaque flacon contient :
Sotatercept............................................................................................................................ 45 mg
Après reconstitution, chaque mL de solution contient 50 mg de sotatercept.
Le sotatercept est une protéine de fusion recombinante homodimérique produite dans les cellules d'ovaires de hamster chinois (CHO) par la technique de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Poudre :
Acide citrique monohydraté (E330)
Citrate trisodique dihydraté (E331)
Polysorbate 80 (E433)
Saccharose.
Solvant :
Eau pour préparations injectables.